

Leucemia Linfoblástica Aguda

Pelo menos uma vez na vida já ouvimos falar em leucemia, seja na ficção, conhecidos que passaram por essa condição e até mesmo nos meios de comunicação mais conhecidos como impressos, televisão e internet.
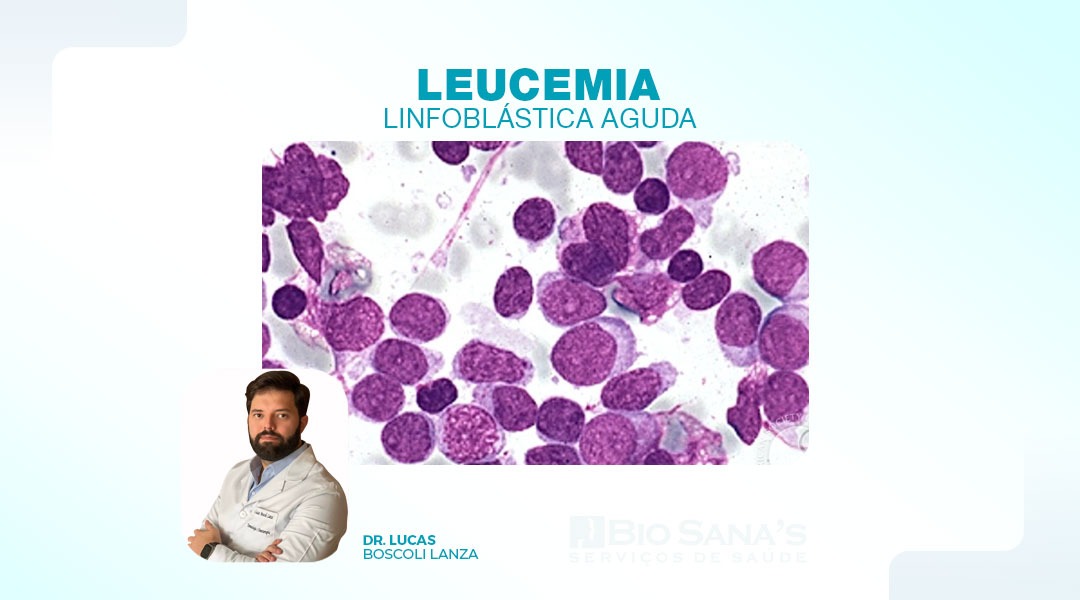
As leucemias são um grupo heterogêneo de neoplasias (doenças oncológicas) que se originam na medula óssea e acometem os precursores hematopoéticos, ou seja, as células que darão origem as hemácias, leucócitos (granulócitos e linfócitos) e plaquetas, tanto a célula precursora hematopoética que ainda não se diferenciou nessas linhagens, quanto suas formas jovens já em processo de maturação inicial. A palavra leucemia deriva-se da união de palavras de origem grega “leukos” (branco) e “haima” (sangue). Quando adicionamos o “sobrenome” aguda, trata-se de um grupo de doenças que tem como via final a produção de células jovens anormais que não atingem a maturação completa e se proliferam de forma clonal (ou seja, produzem mais e mais células tumorais idênticas). Essas células malignas sem a capacidade de amadurecimento e de alto índice proliferativo são os conhecido por muitos como blastos ou, no caso dessa postagem, dos linfoblastos. A origem dessas alterações ocorre na própria medula óssea – nossa grande fábrica de sangue – que está doente e produz os blastos de determinada linhagem a partir de alterações citogenéticas e biomoleculares.
Nesse post vamos nos ater as Leucemias Linfoblásticas Agudas que, a partir desse ponto, serão citadas como LLA. Por se tratar de um câncer que acomete linfócitos produzidos na medula óssea, podem ser didaticamente divididos em LLA de células B e LLA de células T a depender do tipo de linfócito acometido.
A incidência anual, considerando dados mundiais mais atuais, é de 1 a 5 casos a cada 100.000 pessoas por ano e, destes, mais de dois terços dos casos são de linhagem B. As LLA são patologias que ocorrem predominantemente na infância (menores de 6 anos de idade) sendo essa faixa etária responsável por aproximadamente 75% da incidência geral. Um segundo pico de incidência ocorre em adultos acima de 60 anos. A incidência, ainda, é maior na população de origem hispânica e em indivíduos do gênero masculino. Infelizmente não se conhece completamente fatores de risco definitivos, mas há alguma evidência de que, nas formas infantis, possa se seguir de mutações herdadas ainda na fase fetal (como Síndrome de Down, dentre outras) e por infecções (sobretudo pelo HTLV-1) também nesse período e na infância. Em adultos também existem poucos fatores de risco conhecidos, destacando-se a exposição a radiação ionizante e contato com agentes pesticidas. Outro fato interessante, é que raramente a LLA possui padrão de herança familiar, ocorrendo na maioria das vezes como um diagnóstico de novo (“ao acaso”). Sob o conhecimento dessas informações, é de se esperar que não haja ainda uma forma bem validada de rastreio ativo para essa patologia.
A apresentação clínica das LLA é variável, seja a LLA-B quanto a LLA-T, podendo ser oligossintomática e de progressão lenta em semanas a meses, mas também podem ser agressivas, com evolução em poucos dias, situação em que os sintomas são mais exuberantes e até mesmo fatais se não reconhecidos imediatamente. Quando há intenso acometimento intramedular pelos blastos é de se imaginar que a atividade de produção das outras linhagens da medula óssea esteja reduzida, resultando em anemia (fraqueza, dispneia, baixa tolerância a exercício, sonolência etc.), leucopenia (febre e infecções – sejam fúngicas, virais e bacterianas) e plaquetopenia (púrpuras, manchas arroxeadas na pele e sangramentos). Os chamados sintomas B, composto por sudorese noturna, febre e perda ponderal não intencional de mais de 10% do peso corporal nos últimos 6 meses, também podem ocorrer. Além desses sinais e sintomas provocados pelas alterações da homeostase em si, a LLA pode provocar linfadenomegalia (as populares “ínguas”), aumento do fígado e do baço, infiltração em gônadas, glândula suprarrenal, rins e Sistema Nervoso Central (e, pasmem, a infiltração neurológica ocorre em até 20% dos casos) – nestes casos a LLA passa a ser uma entidade conhecida como LLA/linfoma linfoblástico de células B ou T, a depender do linfoblasto envolvido.
O diagnóstico das LLA deve ser excepcionalmente realizado pelo hematologista que, ao suspeitar da patologia com os achados de exames, sobretudo o hemograma, prossegue para a pesquisa citogenética e biomolecular, geralmente através do mielograma, essenciais para confirmação diagnóstica, classificação da LLA e sua estratificação de risco. As informações obtidas com essa pesquisa nos auxiliam na escolha do esquema terapêutico, uma vez disponíveis drogas alvo-dirigidas contra proteínas expressas na superfície celular e contra determinadas mutações genéticas. Diferentemente do que se conhece nas Leucemias Mieloides Agudas (LMA), não há um valor mínimo de blastos requerido para fechar esse diagnóstico, desde que as alterações citogenéticas e biomoleculares sejam compatíveis.
Após o diagnóstico e revisão de todos os dados do paciente e das células malignas podemos classificar o paciente como risco padrão e em alto risco para desfechos graves e recidivas. Fatores de mau prognóstico para a LLA-B são: idade ao diagnóstico menor que 1 ano e maior que 40 anos, contagem de glóbulos brancos maior que 30 mil/uL, cariótipo (perfil genético) complexo, alterações genéticas específicas como o rearranjo KMT2A, IGH e BCR-ABL1 (esse último resultando translocação cromossômica 9;22, resultando no famoso cromossomo Philadelphia) e ausência de resposta a terapia inicial. Para a LLA-T os fatores de mau prognóstico são semelhantes, excluindo-se algumas das mutações que são mais frequentes na LLA-B e adicionando-se o momento em que houve parada de maturação, sendo assim pior quando se trata linfócitos T muito imaturos (como é o caso do “Early T”).
O tratamento das LLA, seja B ou T, não é uma receita pronta – na verdade diria ser algo até artesanal. Ele requer que conheçamos nosso paciente e sua doença: sua idade, comorbidades prévias, funcionalidade, estratificação de risco e tratamentos já realizados. Seja qual fora a variante da LLA, o tratamento deve contemplar as seguintes fases: indução, consolidação, intensificação e manutenção.
No tratamento da LLA-B, em pacientes virgens de tratamento e com boa funcionalidade e reserva orgânica, optamos por protocolos de quimioterapia baseados em estudos (como GRAALL-2005, GRAAPH-2005 (quando há a presença do cromossomo Philadelphia), BFM-2009, dentre outros). Dentre as medicações utilizadas estão os glicocorticoides, Rituximabe, Vincristina, L-Asparaginase, Antraciclinas, Citarabina, Metotrexate, Mercaptopurina e a sempre presente quimioterapia intratecal (lembram-se dos 20% dos casos afetando o sistema nervoso central?). No caso de associação com cromossomo Philadelphia (também chamada LLA Ph+), faz-se necessário o uso de Inibidores de tirosina quinase, como Imatinibe, Dasatinibe, Nilotinibe ou Ponatinibe associados ao esquema quimioterápico padrão. Outra opção, ainda em primeira linha, seria o esquema linear R-HYPER-CVAD que abrange a maioria das drogas citadas previamente. Para a consolidação dos casos de alto risco, preconiza-se o transplante de medula óssea alogênico. Em casos especiais pode ser optado o TMO autólogo, mas pouco utilizado na prática. Já para os casos de risco standard, a quimioterapia padrão pode ser utilizada durante todo o tratamento. Ainda no leque de opções de tratamento da LLA-B, atualmente dispomos de terapias-alvo como os biespecíficos e terapia com células CAR-T, por ora ainda restrita aos tratamentos de recidivas, ou seja, ao menos como segunda linha. Dentre essas drogas são conhecidas o Inotuzumabe-Ozogamicina, o Blinatumomabe (aqui temos uma “quase exceção”, uma vez existem evidências de efetividade em primeira linha em adolescentes e adultos jovens (15 a 39 anos ao diagnóstico). Já sobre o tratamento com células CAR-T, dispomos de produtos comerciais, como Tisagenlecleucel e Axicabtageno Ciloleucel e outros em protocolos de estudos.
Já no tratamento da LLA-T, as opções são mais reduzidas e as respostas ao tratamento são inferiores as obtidas no caso da LLA-B. Aqui podemos utilizar HYPER-CVAD, mas a maioria dos poucos tratamentos disponíveis são também baseados em estudos clínicos. Já para casos refratários, dispomos da Nelarabina. Biespecíficos e terapia com células CAR-T ainda estão em estudo.
Por fim, quando falamos em remissão, essa é muito variável de acordo com a fonte consultada. As taxas de cura são maiores em crianças, podendo chegar a 80-90%. Já em adultos as taxas de remissão podem atingir níveis tão altos quanto 90%, entretanto, em até metade dos casos podem ocorrer recidivas. Já adultos que atingem remissão completa sem sinais de recaída, a taxa pode variar de 40 a 50%.
Independente das taxas de remissão e cura obtidas nos mais diversos estudos clínicos é unânime que diagnósticos mais precoces possuem melhores desfechos. Portanto, ao notar sinais como citados acima, é de suma importância procurar atendimento médico especializado.
Bibliografia utilizada:
- Sebastian G. How I treat newly diagnosed acute lymphoblastic leukemia. Clin Hematol Int. 2024 May 9;6(2):51-61. doi: 10.46989/001c.117026. PMID: 38817308; PMCID: PMC11088446.
- Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet. 2020 Apr 4;395(10230):1146-1162. doi: 10.1016/S0140-6736(19)33018-1. PMID: 32247396.
Texto elaborado por Dr. Lucas Boscoli Lanza, Médico Hematologista especialista em transplante de medula óssea da Equipe BIO SANA’S, Hospital São Camilo Pompéia e IBCC Oncologia (Instituto Brasileiro de Controle do Câncer).
Para saber sobre transplante de medula óssea, basta clicar no link abaixo:
https://www.biosanas.com.br/post/160/quais-as-etapas-do-transplante-de-medula-ossea-tmo



